Understanding atomic and molecular interactions is fundamental to both chemistry and physics, yet it is a labyrinthine challenge fraught with complexities. Atoms, composed of a positively charged nucleus orbited by negatively charged electrons, interact in ways that can be extremely difficult to simulate. When these atoms combine to form molecules, their electrons engage in intricate interactions governed by quantum mechanics. Consequently, the simulation of molecular dynamics becomes one of the most formidable tasks in modern scientific inquiry. The complexity amplifies exponentially as the number of atoms increases, often requiring days or even weeks of computational time on high-performance computing systems.
The challenge primarily lies in accurately solving the Schrödinger equation, which describes an atom’s or molecule’s energy levels and behaviors. For systems larger than a few dozen atoms, the computational resources required quickly outstrip what is practical, limiting progress in fields like drug development and materials science. This scenario necessitates a fresh approach to molecular simulations—enter the realm of machine learning (ML). Researchers recognize that traditional methods are not only inefficient but could also benefit from the advent of innovative computational techniques.
Machine Learning: A Game Changer for Simulations
Recent advancements highlight the transformative potential of machine learning in simplifying and enhancing molecular simulations. Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin, in collaboration with Google DeepMind, have made significant strides by developing a new algorithm that streamlines the process of simulating molecular interactions. This algorithm not only allows for the precise simulation of single and multiple molecules but does so over extended time scales, a feat previously considered nearly unattainable.
Machine learning excels in its ability to predict outcomes based on learned data, sidestepping the need for explicit solutions to the Schrödinger equation. Instead of rigidly generating every variable, the algorithm learns to comprehend the essential dynamics of electron interactions, reducing the computational load while maintaining high accuracy. This paradigm shift offers a glimpse into a future where simulation becomes not just feasible but efficient enough to revolutionize the approach to various scientific inquiries.
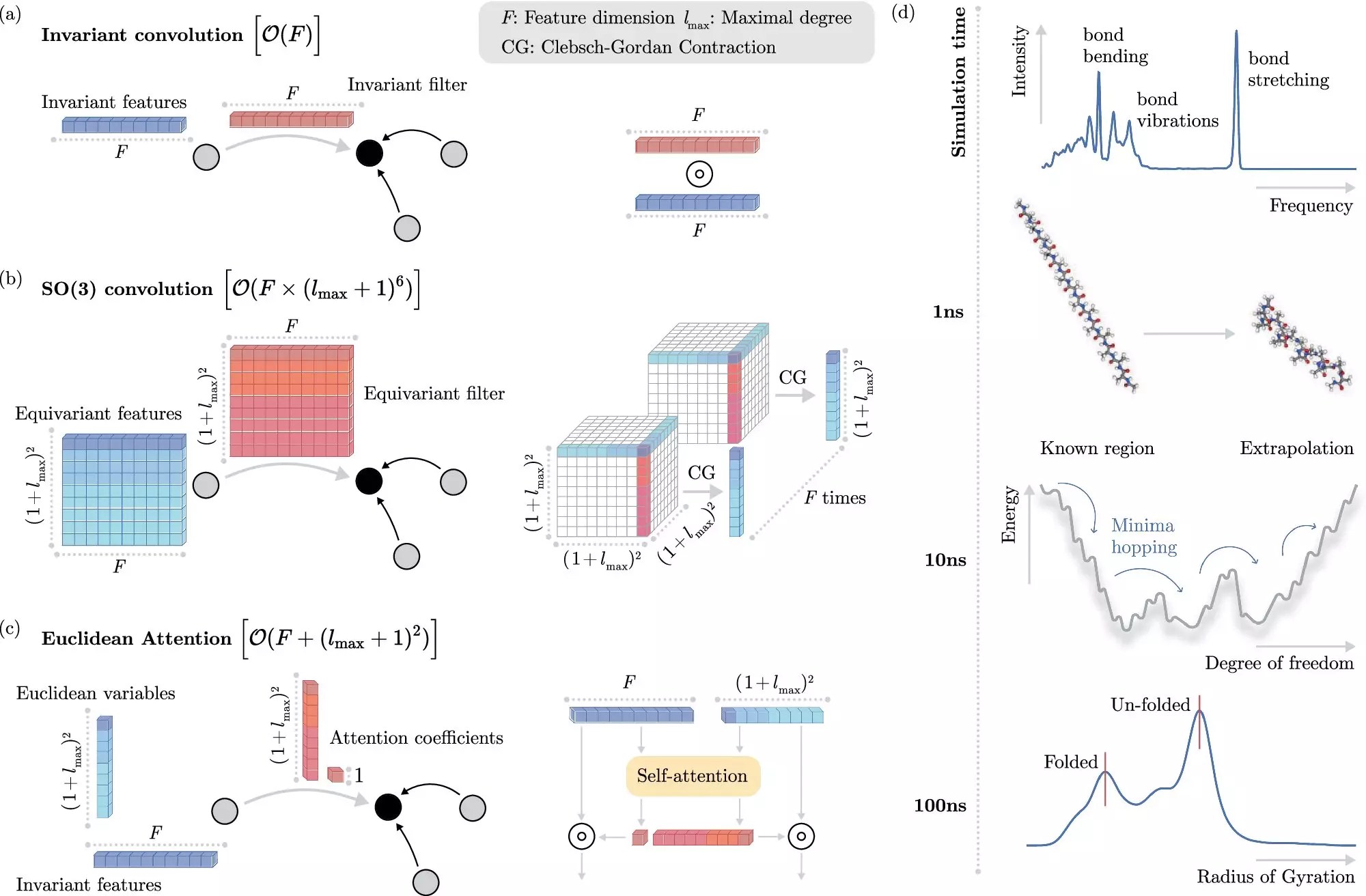
Efficiency through Invariance
One of the most notable aspects of the new algorithm is its innovative treatment of invariances—properties of physical systems that resist change even when the systems themselves are manipulated spatially. Traditional machine learning methods have struggled with efficiently incorporating these invariances, often bogging down the computational process and limiting scalability. However, BIFOLD’s new approach disentangles invariances from the rest of the information about a chemical system right from the start. This shift not only simplifies the learning process but allocates computational resources more effectively, allowing for sophisticated molecular dynamics simulations to be executed in days rather than months.
This efficiency leap has monumental implications. What once required vast computational clusters can now be managed on a mere single computer node. Crucial simulations for understanding the structure and functionality of molecular systems can happen at a speed and scale that was previously unimaginable.
Broader Implications for Drug Development
The multifaceted applications of this novel algorithm in drug development and material science are profoundly promising. For instance, researchers can now predict the interaction between molecules and proteins within the human body with unprecedented accuracy. This transformational capability means that the identification and creation of new drugs could soon be accomplished without the high costs and extensive timelines associated with traditional experimental methods.
One substantial instance of this potential is illustrated by the identification of stable molecular forms of docosahexaenoic acid, a fatty acid integral to brain structure and development. By efficiently scanning thousands of molecular candidates, the researchers showcased the viability of their machine learning method in a real-world application that was infeasible using conventional quantum mechanical approaches.
A Step Towards Future Research
The implications of this research extend beyond mere efficiency; it represents a paradigm shift that intertwines advanced machine learning techniques with the foundational laws of physics. As stated by leading scientists involved in the research, this breakthrough paves the way for addressing long-standing enigmas in computational chemistry. The road ahead involves tackling more intricate systems while ensuring accurate representation of complex, long-range interactions.
The collaborative efforts at BIFOLD and Google DeepMind showcase the potential of interdisciplinary research in solving real-world problems. At a time when efficient, scalable methods are in dire need, this novel machine learning approach stands out as a beacon of innovation, laying the groundwork for future explorations that could redefine molecular dynamics as we know it.
Leave a Reply